Intrinsic point defects and the $n$- and $p$-type dopability of the narrow gap semiconductors GaSb and InSb
Abstract
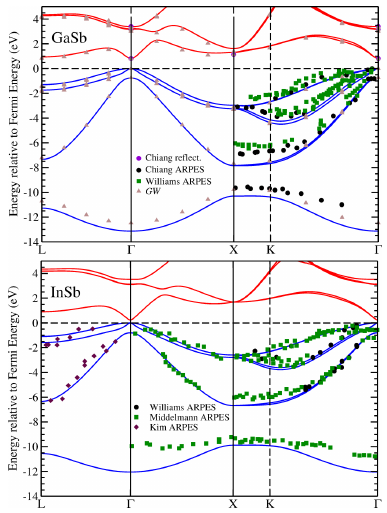
The presence of defects in the narrow-gap semiconductors GaSb and InSb affects their dopability and hence applicability for a range of optoelectronic applications. Here, we report hybrid density functional theory based calculations of the properties of intrinsic point defects in the two systems, including spin orbit coupling effects, which influence strongly their band structures. With the hybrid DFT approach we adopt, we obtain excellent agreement between our calculated band dispersions, structural, elastic and vibrational properties and available measurements. We compute point defect formation energies in both systems, finding that antisite disorder tends to dominate, apart from in GaSb under certain conditions, where cation vacancies can form in significant concentrations. Calculated self-consistent Fermi energies and equilibrium carrier and defect concentrations confirm the intrinsic $n$- and $p$-type behaviour of both materials under anion-rich and anion-poor conditions. Moreover, by computing the compensating defect concentrations due to the presence of ionised donors and acceptors, we explain the observed dopability of GaSb and InSb.
These calculations were on the ‘back burner’ for a while, but ended up providing some insights into the electronic properties of GaSb and InSb, two standard semiconducting materials that not many people have looked into using first electronic structure theory calculations. The defect physics of both are investigated, using plane-wave, hybrid DFT, where the hybrid functional is chosen to reproduce only the band gap of each, but it is shown that the bulk properties of each system are reproduced well.
John Buckeridge
Lecturer in Energy Engineering and Materials Devices
Materials physicist working at the School of Engineering - Division of Electrical and Electronic Engineering, London South Bank University.