Investigating defects in wide-gap nitrides and oxides.
Software
Available codes
SC-FERMI
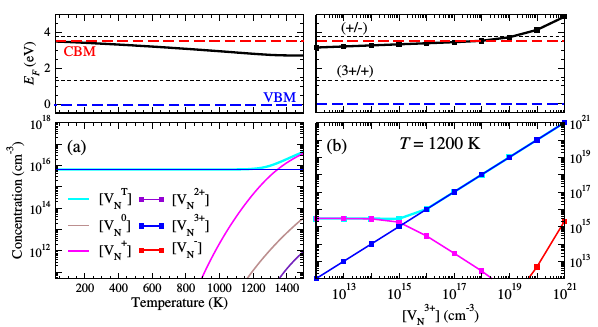
Results for N vacancies in GaN (see Reference).
If you know the formation energies for a set of defects in a material and you know the density of states, given the constraint of charge neutrality, you can work out the self-consistent Fermi energy. SC-FERMI is a code designed to do this for you. It reads in the formation energies and density of states and outputs equilibrium defect and carrier concentrations, as well as the Fermi energy, for a given temperature. You can also set particular defect concentrations to remain constant, therefore modelling the situation where some defects are ‘frozen in’ by kinetic barriers. Follow the links to find out more.
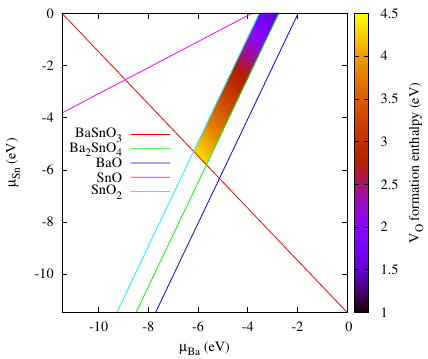
The Chemical Potential Limits Analysis Program (CPLAP!) is designed to determine the thermodynamical stability of a material, and, if it is stable, to determine the ranges of the constituent elements’ chemical potentials within which it is stable, in comparison with competing phases and the elemental forms. When determining defect formation energies in a material, CPLAP allows you to calculate the allowed ranges of chemical potentials at which the defects can form. These ranges are tedious to calculate when you have a multiternary system, so having an automated procedure can be quite helpful. CPLAP also produces plotting files if the space spanned is 2D or 3D. There are various features, please follow the links to obtain the code or read the relevant citation.